- Home
- Entre falhas inatas e adquiridas na imunidade
Entre falhas inatas e adquiridas na imunidade
As complicações decorrentes da imunodeficiência e o tipo de agente etiológicoNos primeiros anos de vida, o sistema imune é exposto aos mais variados antígenos pela primeira vez, montando uma resposta e adquirindo memória imunológica. É natural, portanto, que a população pediátrica seja mais vulnerável às infecções, bem como que estas se manifestem de forma recorrente em tal faixa etária.
Principais imunodeficiências na população infantil

Imunodeficiência comum variável pode ocorrer em qualquer idade
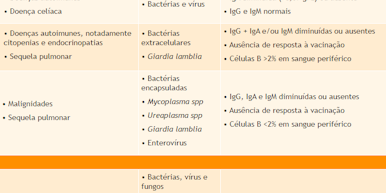
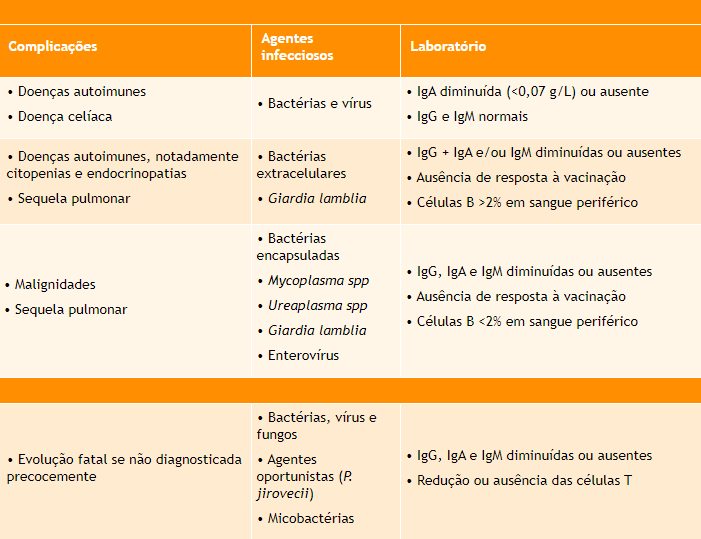
Considerada um diagnóstico de exclusão, a imunodeficiência comum variável (ICV) abrange um grupo heterogêneo de doenças semelhantes, porém com defeitos moleculares distintos, caracterizando-se pela presença de infecções recorrentes, pela redução dos níveis séricos de IgG e de mais uma classe de imunoglobulina, pelo menos, e por ausência na produção de anticorpos específicos após exposição natural a um antígeno ou estímulo com vacina.
Nos pacientes com ICV, os sintomas podem começar em qualquer idade, especialmente na segunda e terceira décadas de vida. Há destaque para as infecções gastrointestinais, nas quais a Giardia lamblia configura um agente etiológico relevante, e para as sinopulmonares, causadas por bactérias extracelulares. Nesses casos, com frequência o quadro evolui para sequelas pulmonares, mesmo diante de tratamento adequado. Apesar de a característica principal da doença ser a deficiência de anticorpos, existe a possibilidade de alguns pacientes apresentarem também comprometimento da imunidade celular.
Os portadores dessa imunodeficiência têm ainda maior suscetibilidade a afecções inflamatórias e autoimunes, como enteropatias, inflamação granulomatosa e citopenias, assim como a neoplasias, principalmente hematológicas, o que costuma ser bem caracterizado e pode até mesmo ser a primeira manifestação da ICV.
Do que desconfiar na infância?
envolvido dão pistas importantes para o diagnóstico clínico.
Contudo, cabe lembrar que, apesar de a maioria das crianças que adoecem com frequência ter imunidade normal, algumas podem realmente apresentar uma imunodeficiência de base e precisam ser reconhecidas. Conheça as principais hipóteses a suspeitar nesse grupo.

Deficiência congênita do complemento compromete a imunidade inata
Formado por um conjunto de proteínas séricas, o sistema complemento é parte da resposta imune inata e participa da importante função de impedir a instalação das infecções, promovendo a destruição de microrganismos e a depuração de imunocomplexos e estimulando a inflamação.
A deficiência de componentes iniciais da via clássica (C1q, C2 e C4) está mais frequentemente associada a doenças autoimunes, em especial ao lúpus eritematoso sistêmico. Já a deficiência primária de C3 e dos componentes tardios (C5 a C9) da via comum do sistema complemento está entre as imunodeficiências mais raras e se relaciona com uma maior vulnerabilidade a processos infecciosos, sendo as bactérias encapsuladas, como a Neisseria meningitidis, o Streptococcus pneumoniae e o Haemophilus influenzae, os patógenos mais comumente envolvidos.
Os defeitos na via do complemento podem ser inicialmente investigados por meio de ensaios hemolíticos para avaliação da via clássica (CH 50) e, se necessário, da via alternativa (AP 50). Os exames fornecem uma ideia da integridade funcional da cascata do complemento. Caso haja anormalidade, devem ser dosados os componentes individuais apropriados, de acordo com o cenário clínico-laboratorial.
• Autoimunidade e alta suscetibilidade para infecções
• Doenças autoimunes na infância (em idade precoce e no sexo masculino)
• Autoimunidade associada a manifestações atópicas ou similares
• Múltiplas doenças autoimunes em qualquer idade
• História familiar de imunodeficiência primária
 01 de junho de 2014
01 de junho de 2014Outros destaques dessa edição